Researchers develop first human-relevant microphysiological model of neuromuscular circuits for Duchenne muscular dystrophy
A team of researchers from Dr Yung-Yao Lin’s group at the Blizard Institute, in collaboration with Dr Ivo Lieberam at King's College London, have developed the first light-controllable neuromuscular model for Duchenne muscular dystrophy (DMD) using bioengineering techniques, pluripotent stem cells, and optogenetics – a neuromodulation method that uses genetically engineered “light sensors” to control the activities of individual neurons in living tissue.
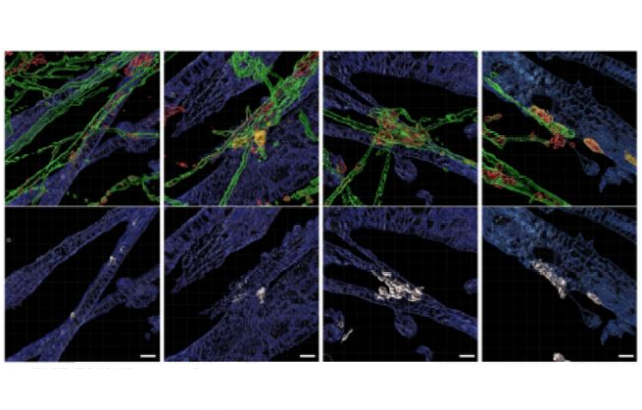
Representative 3D reconstruction images of human NMJs in (L-R) DMD-R3381X, DMD-R3381X + gentamicin, DMD-R3381X + SB-431542, and CORR-R3381X neuromuscular circuit cocultures.
The study, published today in Science Advances, was co-funded by Barts Charity, with patient charities Action Duchenne and Duchenne Parent Project providing funding to the Yung-Yao Lin lab, and the Medical Research Council (MRC) and the Wellcome Trust providing funding to the team at King's.
DMD is caused by mutations in the dystrophin gene. It is fatal and the most common neuromuscular disorder in childhood. DMD patients suffer from progressive skeletal muscle weakness and wasting that leads to eventual loss of ambulation with poor quality of life and reduced life expectancy. Despite some existing drugs, there is still no cure or effective treatment for DMD.
In this study, the researchers developed the first light-controllable microphysiological model for Duchenne muscular dystrophy (DMD) with patient-derived and gene-corrected neuromuscular circuits in fabricated microdevices recapitulating key features of muscle-nerve connectivity in the human body.
The formation of neuromuscular circuits is critical for generating voluntary movement, in which skeletal muscle contraction is induced by motor neurons through neurotransmitter release at neuromuscular junctions (NMJs). Progress in finding a cure for neuromuscular diseases has been hampered by the lack of human-relevant microphysiological models for studying neuromuscular circuits in healthy and disease conditions, as well as for assessing the efficacy of therapeutic strategies.
The study identified abnormal gene expression in DMD myofibers, including key genes of the NMJ and showed that NMJ volumes and optogenetic motor neuron-stimulated myofiber contraction are compromised in DMD neuromuscular circuits.
The researchers also established a high content imaging-compatible, 96-well human neuromuscular circuit co-culture assay that can simultaneously distinguish drugs with different mechanisms of action for restoring NMJ defects. For the first time, they show that the motor function of DMD neuromuscular circuits can be remarkably restored by pharmacological inhibition of TGFβ signalling (SB-431542). This is an unexpected finding beyond the role of TGFβ signalling in myofiber homeostasis.
Dr Yung-Yao Lin from the Centre for Genomics and Child Health at the Blizard Institute said: “Our study provides a new human-relevant microphysiological model for investigating NMJ defects in DMD and evaluating candidate drugs and suggests enhancing neuromuscular connectivity may be an effective therapeutic strategy.”
The experimental approach is applicable to other neuromuscular disorders, such as amyotrophic lateral sclerosis (ALS). The findings hold promise to offer improved translatability and reduce the use of animals in research.
More information
- Find out more about the Centre for Genomics and Child Health at the Blizard Institute.
- Find out more about the Centre for Predictive in vitro Models at Barts and The London School of Medicine and Dentistry, Queen Mary University of London.
Related news
- Q&A: New patient-derived iPSC model for congenital muscular dystrophies demonstrates feasibility of identifying novel drug therapies – Monday 30 September 2019
- New research funding brings new hope for Duchenne Muscular Dystrophy – Wednesday 9 May 2018