![[eta]](../greek/ETA.GIF) -symbol ("eta")
-symbol ("eta")
Contents
ET-value; + educt; effective charge; effective molarity (or effective concentration); eighteen-electron rule; electrocyclic reaction; electrofuge; electromeric effect; electron acceptor; electron affinity; electronation; electron attachment; electron configuration; electron-deficient bond; electron density; electron detachment; electron donor; + electron-donor-acceptor complex; electronegativity; electroneutrality principle; electronic effect of substituents: symbols and signs; electron-pair acceptor; electron-pair donor; electron transfer; electron-transfer catalysis; electrophile, electrophilic; electrophilicity; element effect; elementary reaction; elimination; enantiomer; enantioselectivity; encounter complex; encounter-controlled rate; ene reaction; energy of activation (Arrhenius energy of activation; activation energy); energy profile; enforced concerted mechanism; enophile; entering group; enthalpy of activation (standard entalpy of activation); entropy of activation, (standard entropy of activation); epimer; epimerization; equilibrium, chemical; equilibrium control; equilibrium ET-value; + educt; isotope effect; excess acidity; excimer; exciplex; excited state; extended Hammett equation; external return; extrusion transformation; -symbol ("eta")
See Dimroth-Reichardt ET parameter, Z-value.
Term used mainly in the German literature for starting material (reactant). It should be avoided in English, because there it means "something that comes out" and not "something that goes in". The German use of the term is in fact also incorrect.
Change in effective charge is a quantity obtained by comparison of the polar effect of substituents on the free energies of rate or equilibrium processes with that on a standard ionization equilibrium. Provided the effective charge on the states in the standard equilibrium are defined, then it is possible to measure effective charges for states in the reaction or equilibrium under consideration. WILLIAMS (1984, 1992).
effective molarity (or effective concentration)
The ratio of the first-order rate constant of an intramolecular reaction involving two functional groups within the same molecular entity to the second-order rate constant of an analogous intermolecular elementary reaction. This ratio has the dimension of concentration. The term can also apply to an equilibrium constant. See also intramolecular catalysis.
An electron-counting rule to which an overwhelming majority of stable diamagnetic transition metal complexes adhere. The number of nonbonded electrons at the metal plus the number of electrons in the metal-ligand bonds should be 18. The 18 electron rule in transition metal chemistry is a full analogue of the "Lewis octet rule".
A molecular rearrangement that involves the formation of a sigma bond between the termini of a fully conjugated linear pi-electron system (or a linear fragment of a pi-electron system) and a decrease by one in the number of pi bonds, or the reverse of that process. For example:
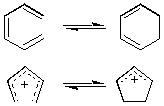
The stereochemistry of such a process is termed "conrotatory" or antarafacial if the substituents at the interacting termini of the conjugated system both rotate in the same sense, e.g.
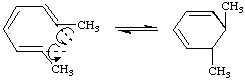
or "disrotatory" (or suprafacial) if one terminus rotates in a clockwise and the other in a counter-clockwise sense, e.g.
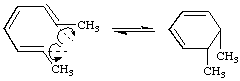
See also pericyclic reaction.
A leaving group that does not carry away the bonding electron pair. For example, in the nitration of benzene by NO2+, H+ is the electrofuge. The adjective of electrofuge is electrofugal. See also electrophile, nucleofuge.
A molecular polarizability effect occurring by an intramolecular electron displacement (sometimes called the "conjugative mechanism" and, previously, the "tautomeric mechanism") characterized by the substitution of one electron pair for another within the same atomic octet of electrons. It can be indicated by curved arrows symbolizing the displacement of electron pairs, as in
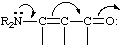
which represents the hypothetical electron shift
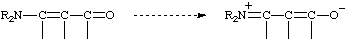
The term has been deemed obsolescent or even obsolete (see mesomeric effect, resonance effect). It has long been custom to use phrases such as "enhanced substituent resonance effect" which imply the operation of the electromeric effect, without using the term, and various modern theoretical treatments parametrize the response of substituents to "electronic demand", which amounts to considering the electromeric effect together with the inductomeric effect. EHRENSON, BROWNLEE and TAFT (1973); TAFT and TOPSOM (1987); CHARTON (1987).
(1) A substance to which an electron may be transferred; for example 1,4-dinitrobenzene or the dication 1,1'-dimethyl-4,4'-bipyridyldiium .
+ (2) A Lewis acid. This usage is discouraged.
The energy released when an additional electron (without excess energy) attaches itself to a molecular entity (usually an electrically neutral molecular entity). (The direct measurement of this quantity involves molecular entities in the gas phase.)
See electron attachment, reduction.
The transfer of an electron to a molecular entity, resulting in a molecular entity of (algebraically) increased negative charge. (It is not an attachment, as defined in this Glossary.)
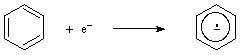
See also oxidation (1), reduction.
See configuration (electronic).
A single bond between adjacent atoms that is formed by less than two electrons, as in B2H6:
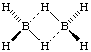
The B-H-B bonds are called a "two-electron three-centre bonds".
If P(x,y,z) dx dy dz is the probability of finding an electron in the volume element dx dy dz at the point of a molecular entity with coordinates x,y,z, then P(x,y,z) is the electron density at this point. For many purposes (e.g., X-ray scattering, forces on atoms) the system behaves exactly as if the electrons were spread out into a continuously distributed charge. The term has frequently been wrongly applied to negative charge population. See also charge density.
The reverse of an electron attachment.
(1) A molecular entity that can transfer an electron to another molecular entity, or to the corresponding chemical species.
+ (2) A Lewis base. This use is discouraged.
+ electron-donor-acceptor complex
A term sometimes employed instead of charge-transfer complex or Lewis adduct. See also adduct, coordination.
A measure of the power of an atom or a group of atoms to attract electrons from other parts of the same molecular entity. The concept has been quantified by a number of authors, including especially Pauling and Mulliken. See ATKINS (1974).
The principle expresses the fact that all pure substances carry a net charge of zero.
electronic effect of substituents: symbols and signs
The inductive effect has universally been represented by the symbol I. This is now commonly taken to include both through-bonds and through-space transmission, but I is also used specifically for through-bonds transmission; through-space transmission is then symbolized as F (for field effect). The symbols for the influence of substituents exerted through electron delocalization have variously been M (mesomeric), E (electromeric), T (tautomeric), C (conjugative), K (konjugativ), and R (resonance). Since the present fashion is to use the term resonance effect, R is the most commonly used symbol, although M is still seen quite often.
Both the possible sign conventions are in use. The Ingold sign convention associates electronegativity (relative to hydrogen atom) with a negative sign, electropositivity with a positive sign. Thus the nitro group is described as electron- withdrawing by virtue of its - I and - M effects; chloro is described as a - I, +M substituent, etc. For correlation analysis and linear free-energy relationships this convention has been found inconvenient, for it is in contradiction to the sign convention for polar substituent constants (![[sigma]](../greek/SIGMA.GIF) -constants). Authors concerned with these fields often avoid this contradiction by adopting the opposite sign convention originally associated with Robinson, for electronic effects. This practice is almost always associated with the use of R for the electron delocalization effect: thus the nitro group is a +I, +R substituent; chloro a +I, - R substituent, etc.
-constants). Authors concerned with these fields often avoid this contradiction by adopting the opposite sign convention originally associated with Robinson, for electronic effects. This practice is almost always associated with the use of R for the electron delocalization effect: thus the nitro group is a +I, +R substituent; chloro a +I, - R substituent, etc.
A synonym for Lewis acid.
A synonym for Lewis base.
The transfer of an electron from one molecular entity to another, or between two localized sites in the same molecular entity. See also inner sphere (electron transfer), outer sphere (electron transfer), Marcus equation.
The term indicates a sequence of reactions such as shown in equations (1)-(3), leading from A to B:
A. - ![]() B. - (2)
B. - (2)
B. - + A ![]() B + A. - (3)
B + A. - (3)
An analogous sequence involving radical cations (A.+, B.+) is also observed.
The most notable example of electron-transfer catalysis is the SRN1 (or T+DN+AN) reaction of aromatic halides.
The term has its origin in a suggested analogy to acid-base catalysis, with the electron instead of the proton. However, there is a difference between the two catalytic mechanisms, since the electron is not a true catalyst, but rather behaves as the initiator of a chain reaction. "Electron-transfer induced chain reaction" is a more appropriate term for the situation described by equations (1)-(3). EBERSON (1987).
An electrophile (or electrophilic reagent) is a reagent that forms a bond to its reaction partner (the nucleophile) by accepting both bonding electrons from that reaction partner.
An "electrophilic substitution reaction" is a heterolytic reaction in which the reagent supplying the entering group acts as an electrophile. For example
Electrophilic reagents are Lewis acids. "Electrophilic catalysis" is catalysis by Lewis acids.
The term "electrophilic" is also used to designate the apparent polar character of certain radicals, as inferred from their higher relative reactivities with reaction sites of higher electron density. See also electrophilicity.
(1) The property of being electrophilic (see electrophile).
(2) The relative reactivity of an electrophilic reagent. (It is also sometimes referred to as "electrophilic power".) Qualitatively, the concept is related to Lewis acidity. However, whereas Lewis acidity is measured by relative equilibrium constants, electrophilicity is measured by relative rate constants for reactions of different electrophilic reagents towards a common substrate (usually involving attack at a carbon atom). See also nucleophilicity.
The ratio of the rate constants of two reactions that differ only in the identity of the element of the atom in the leaving group, e.g., kBr/kCl. As for isotope effects, a ratio of unity is regarded as a "null effect".
A reaction for which no reaction intermediates have been detected or need to be postulated in order to describe the chemical reaction on a molecular scale. An elementary reaction is assumed to occur in a single step and to pass through a single transition state. IUPAC CHEMICAL KINETICS (1981). See also stepwise reaction.
The reverse of an addition reaction or transformation.
In an elimination two groups (called eliminands) are lost most often from two different centres (1/2/elimination or 1/3/elimination, etc.) with concomitant formation of an unsaturation in the molecule (double bond, triple bond) or formation of a new ring.
If the groups are lost from a single centre (![[alpha]](../greek/ALPHA.GIF) -elimination, 1/1/elimination) the resulting product is a carbene or a "carbene analogue". See also -elimination.
-elimination, 1/1/elimination) the resulting product is a carbene or a "carbene analogue". See also -elimination.
One of a pair of molecular entities which are mirror images of each other and non-superimposable.
See stereoselectivity.
A complex of molecular entities produced at an encounter-controlled rate, and which occurs as an intermediate in a reaction mechanism. When the complex is formed from two molecular entities it is called an "encounter pair". A distinction between encounter pairs and (larger) encounter complexes may be relevant in some cases, e.g. for mechanisms involving pre-association.
A rate of reaction corresponding to the rate of encounter of the reacting molecular entities. This is also known as "diffusion-controlled rate" since rates of encounter are themselves controlled by diffusion rates (which in turn depend on the viscosity of the medium and the dimensions of the reactant molecular entities).
For a bimolecular reaction between solutes in water at 25 oC an encounter-controlled rate is calculated to have a second-order rate constant of about 1010 dm3 mol-1 s-1.
See also microscopic diffusion control.
The addition of a compound with a double bond having an allylic hydrogen (the "ene") to a compound with a multiple bond (the "enophile") with transfer of the allylic hydrogen and a concomitant reorganization of the bonding, as illustrated below for propene (the "ene") and ethene (the "enophile"). The reverse is a "retro-ene" reaction.
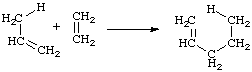
energy of activation (Arrhenius energy of activation; activation energy), Ea (SI unit: kJ mol-1)
An operationally defined quantity expressing the dependence of a rate constant on temperature according to
![[partial differential]](../greek/PARDIF.GIF) ln k /T)p
ln k /T)pas derived from the "Arrhenius equation", k = A exp(-Ea/RT), where A (SI unit: as for the corresponding rate constant) is termed the "pre-exponential factor". See also enthalpy of activation.
See Gibbs energy diagram, potential-energy profile.
Variation of reaction parameters in a series of reactions proceeding in non-concerted steps may lead to a situation, where the putative intermediate will possess a lifetime shorter than a bond vibration, so that the steps become concerted. The transition state structure will lie on the coordinate of the More O'Ferrall-Jencks diagram leading to that of the putative intermediate.
See ene reaction.
An atom or group that forms a bond to what is considered to be the main part of the substrate during a reaction. For example: the attacking nucleophile in a bimolecular nucleophilic substitution reaction.
enthalpy of activation (standard entalpy of activation), ![[Delta]](../greek/DDELTA.GIF)
![[double dagger]](../greek/DDAGGER.GIF) Ho (SI unit: kJ mol-1)
Ho (SI unit: kJ mol-1)
The standard enthalpy difference between the transition state and the ground state of the reactants at the same temperature and pressure. It is related to the temperature coefficient of the rate constant according to the equation:
H = RT2( ln k/T)p - RT = Ea - RT
= -R(ln(k/T)/(1/T))p
where Ea is the energy of activation, providing that the rate constants for reactions other than first-order reactions are expressed in temperature-independent concentration units (e.g., mol dm-3, measured at a fixed temperature and pressure). If lnk is expressed as
then
H = -aR + (c - 1)RT + dRT2.If enthalpy of activation and entropy of activation are assumed to be temperature independent, then
H = -aR
If the concentration units are mol dm-3, the true and apparent enthalpies of activation differ by (n - 1)/(RT2), where n is the order of reaction and the thermal expansivity. See also entropy of activation, Gibbs energy of activation.
entropy of activation, (standard entropy of activation), So (SI unit: J mol-1 K-1)
The standard entropy difference between the transition state and the ground state of the reactants, at the same temperature and pressure.
It is related to the Gibbs energy of activation and enthalpy of activation by the equations
S = (H -G)/T
= H/T - R ln (kB/h) + R ln (k/T)
or, if ln k is expressed as ln k = a/T + b + c ln T + dT,
S = R [b - ln (kB/h) + (c - 1)(1 + ln T) + 2 dT]
provided that rate constants for reactions other than first-order reactions are expressed in temperature-independent concentration units (e.g., mol dm-3, measured at a fixed temperature and pressure). The numerical value of S depends on the standard state (and therefore on the concentration units selected). If entropy of activation and enthalpy of activation are assumed to be temperature-independent,
S = R[b - ln(kB/h)]Strictly speaking, the quantity defined is the entropy of activation at constant pressure from which the entropy of activation at constant volume can be deduced.
The information represented by the entropy of activation may alternatively be conveyed by the pre-exponential factor A (see energy of activation).
A diastereoisomer that has the opposite configuration at only one of two or more tetrahedral "stereogenic" centres present in the respective molecular entity. IUPAC STEREOCHEMICAL GLOSSARY (1993).
Interconversion of epimers by reversal of the configuration at one of the "stereogenic" centres.
Reversible processes (processes which may be made to proceed in the forward or reverse direction by the (infinitesimal) change of one variable, ultimately reach a point where the rates in both directions are identical, so that the system gives the appearance of having a static composition at which the Gibbs energy, G, is a minimum. At equilibrium the sum of the chemical potentials of the reactants equals that of the products, so that
Gr = Gr0 + RT ln K = 0
Gr0 = -RT ln K
The equilibrium constant, K, is given by the mass-law effect.
See isotope effect.
See Dimroth-Reichardt ET parameter, Z-value.
See Bunnett-Olsen equations, Cox-Yates equation.
An excited dimer, "non-bonding" in the ground state. For example, a complex formed by the interaction of an excited molecular entity with a ground state counterpart of this entity. IUPAC PHOTOCHEMICAL GLOSSARY (1992). See also exciplex.
An electronically excited complex of definite stoichiometry, "non-bonding" in the ground state. For example, a complex formed by the interaction of an excited molecular entity with a ground state counterpart of a different structure. IUPAC PHOTOCHEMICAL GLOSSARY (1992). See also excimer.
State of a system with energy higher than that of the ground state. This term is most commonly used to characterize a molecule in one of its electronically excited states, but can also refer to vibrational and/or rotational excitation in the electronic ground state. IUPAC ANALYTICAL CHEMISTRY (1982).
This term applies in a general way to any multiparametric extension of the Hammett equation. It is sometimes used specifically for a form of dual substituent-parameter equation in which the actual value of the correlated property P under the influence of the substituent X is used, rather than the value relative to that for X = H. An intercept term h corresponding to the value of P for X = H is introduced, e.g.
I + ![[beta]](../greek/BETA.GIF) R + h
R + hThe equation may be applied to systems for which the inclusion of further terms to represent other effects, e.g. steric, is appropriate. CHAPMAN and SHORTER (1972, 1978); CHARTON (1987).
See ion-pair return.
A transformation in which an atom or group Y connected to two other atoms or groups X and Z is lost from a molecule, leading to a product in which X is bonded to Z, e.g.,
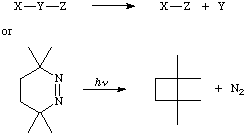
The reverse of an extrusion is called insertion. See cheletropic reaction.
See hapto.
ATKINS, P. W. (1974), "Quanta: a Handbook of Concepts", Clarendon Press, Oxford.
CHARTON, M. (1984), J. Org. Chem., 49, 1997-2001.
CHARTON, M. (1987), Progr. Phys. Org. Chem., 16, 287-315.
EBERSON, L. (1987), "Electron Transfer Reactions in Organic Chemistry", Springer, Berlin.
EHRENSON, S., BROWNLEE, R. T. C., and TAFT, R. W. (1973), Progr. Phys. Org. Chem., 10, 1-80.
*IUPAC STEREOCHEMICAL TERMINOLOGY (1993). IUPAC: Organic Chemistry Division: Basic Terminology of Stereochemistry. IDCNS and public review. Now published as Basic Terminology of Stereochemistry (IUPAC Recommendations 1996) in Pure Appl. Chem., 68, 2193-2222 (1996).
TAFT, R. W., Jr., and TOPSOM, R. D. (1987), Progr. Phys. Org. Chem., 16, 1-83.
WILLIAMS, A. (1984), Acc. Chem. Res., 17, 425-430.
WILLIAMS, A. (1992), Adv. Phys. Org. Chem., 27, 1-55.
Return to home page for Glossary of terms used in Physical Organic Chemistry.