Contents
colligation; common-ion effect (on rates); compensation effect; complementary binding sites; complex; composite reaction; comproportionation; concerted process; condensation reaction; configuration (electronic); configuration (molecular); conformation; conjugate acid-base pair; conjugated system, conjugation; conjugative mechanism; connectivity; constitution; conrotatory; contact ion pair; contributing structure; coordinate covalence (coordinate link); coordination; coordination number; coordinatively saturated; coordinatively unsaturated; coronate; correlation analysis; cosphere; coupling constant (spin-spin coupling constant); covalent bond; Cox-Yates equation; critical micelle concentration (cmc); cross-conjugation; crown; cryptand; Curtin-Hammett principle; cybotactic region; cyclization; cycloaddition; cycloelimination; + cycloreversion
The formation of a covalent bond by the combination or recombination of two radicals (the reverse of unimolecular homolysis). For example:
A reduction in the rate of certain reactions of a substrate RX in solution [by a path that involves a pre-equilibrium with formation of R+ (or R-) ions as reaction intermediates] caused by the addition to the reaction mixture of an electrolyte solute containing the "common ion" X- (or X+). For example, the rate of solvolysis of diphenylmethyl chloride in acetone-water is reduced by the addition of salts of the common ion Cl- which causes a decrease in the quasi-equilibrium concentration of the diphenylmethyl cation in the scheme:
![[reversible arrows]](POgif/CHEM11.GIF) Ph2CH+ + Cl- (free ions, not ion pairs)
Ph2CH+ + Cl- (free ions, not ion pairs)
Ph2CH+ + OH2 ![[arrow]](POgif/CHEM2.GIF) Ph2CHOH + H+(solvated)
Ph2CHOH + H+(solvated)
This phenomenon is a direct consequence of the mass-law effect on ionization equilibria in electrolytic solution.
More generally, the common-ion effect is the influence of the "common ion" on the reactivity due to the shift of the dissociation equilibrium. It may also lead to an enhancement of the rate of reaction.
In a considerable number of cases plots of ![[Delta]](../greek/DDELTA.GIF)
![[double dagger]](../greek/DDAGGER.GIF) SH, for a series of reactions, e.g. for a reaction in a range of different solvents, are straight lines of approximately unit slope. Therefore, the terms H and SG = H - SH or S
SH, for a series of reactions, e.g. for a reaction in a range of different solvents, are straight lines of approximately unit slope. Therefore, the terms H and SG = H - SH or S
See binding site.
A molecular entity formed by loose association involving two or more component molecular entities (ionic or uncharged), or the corresponding chemical species. The bonding between the components is normally weaker than in a covalent bond.
The term has also been used with a variety of shades of meaning in different contexts: it is therefore best avoided when a more explicit alternative is applicable. In inorganic chemistry the term "coordination entity" is recommended instead of "complex" (IUPAC INORGANIC NOMENCLATURE (1990). For the different usage of "complex" in inorganic chemistry, see IUPAC INORGANIC RULES (1970); Rule 2.24. See also activated complex, adduct, charge transfer complex, electron-donor-acceptor complex, encounter complex, inclusion complex, ![[sigma]](../greek/SIGMA.GIF) -adduct,
-adduct, ![[pi]](../greek/PI.GIF) -adduct, transition state.
-adduct, transition state.
A chemical reaction for which the expression for the rate of disappearance of a reactant (or rate of appearance of a product) involves rate constants of more than a single elementary reaction. Examples are "opposing reactions" (where rate constants of two opposed chemical reactions are involved), "parallel reactions" (for which the rate of disappearance of any reactant is governed by the rate constants relating to several simultaneous reactions to form different respective products from a single set of reactants), and stepwise reactions.
The reverse of disproportionation. The term "symproportionation" is also used. HARTMANNS, KLENKE, and METZGER (1986).
Two or more primitive changes are said to be concerted (or to constitute a concerted process) if they occur within the same elementary reaction. Such changes will normally (though perhaps not inevitably) be "energetically coupled". (In the present context the term "energetically coupled" means that the simultaneous progress of the primitive changes involves a transition state of lower energy than that for their successive occurrence.) In a concerted process the primitive changes may be synchronous or asynchronous.
See also bifunctional catalysis, potential energy (reaction) surface.
A (usually stepwise) reaction in which two or more reactants (or remote reactive sites within the same molecular entity) yield a single main product with accompanying formation of water or of some other small molecule, e.g. ammonia, ethanol, acetic acid, hydrogen sulfide.
The mechanism of many condensation reactions has been shown to comprise consecutive addition and elimination reactions, as in the base-catalysed formation of (E)-but-2-enal (crotonaldehyde) from acetaldehyde, via 3-hydroxybutanal (aldol). The overall reaction in this example is known as the aldol condensation.
The term is sometimes also applied to cases where the formation of water or another simple molecule does not occur, as in "benzoin condensation".
A distribution of the electrons of an atom or a molecular entity over a set of one-electron wavefunctions called orbitals, according to the Pauli principle. From one configuration several states with different multiplicities may result. For example, the ground electronic configuration of the oxygen molecule (O2) is
g2, 1u2, 2g2, 2u2, 1u4, 3g2, 1g2resulting in the
![[Sigma]](../greek/SSIGMA.GIF) g, 1g, and 3g+ multiplets
g, 1g, and 3g+ multipletsIUPAC PHOTOCHEMICAL GLOSSARY (1992).
In the context of stereochemistry, the term is restricted to the arrangements of atoms of a molecular entity in space that distinguishes stereoisomers, the isomerism of which is not due to conformational differences. IUPAC STEREOCHEMICAL TERMINOLOGY (1993).
The spatial arrangements of atoms affording distinction between stereoisomers which can be interconverted by rotation about formally single bonds. Some authorities extend the term to include inversion at trigonal bipyramidal centres and other "polytopal rearrangements". IUPAC STEREOCHEMICAL TERMINOLOGY (1993).
The Brønsted acid BH+ formed on protonation of a base B is called the conjugate acid of B, and B is the conjugate base of BH+. (The conjugate acid always carries one unit of positive charge more than the base, but the absolute charges of the species are immaterial to the definition.) For example: the Brønsted acid HCl and its conjugate base Cl- constitute a conjugate acid-base pair.
conjugated system, conjugation
In the original meaning a conjugated system is a molecular entity whose structure may be represented as a system of alternating single and multiple bonds: e.g.
![[triple bond]](../greek/TRIPLEB.GIF) N
NIn such systems, conjugation is the interaction of one p-orbital with another across an intervening sigma bond in such structures. (In appropriate molecular entities d-orbitals may be involved.) The term is also extended to the analogous interaction involving a p-orbital containing an unshared electron pair, e.g.
See also delocalization, homoconjugation, resonance.
See electronic effect.
In a chemical context, the information content of a line formula, but omitting any indication of bond multiplicity.
The description of the identity and connectivity (and corresponding bond multiplicities) of the atoms in a molecular entity (omitting any distinction from their spatial arrangement).
See ion pair.
The definition is based on the valence-bond formulation of the quantum mechanical idea of the wavefunction of a molecule as composed of a linear combination of wavefunctions, each representative of a formula containing bonds that are only single, double or triple with a particular pairing of electron spins. Each such formula represents a contributing structure, also called "resonance structure" to the total wavefunction, and the degree to which each contributes is indicated by the square of its coefficient in the linear combination. The contributing structures, also called "canonical forms", themselves thus have a purely formal significance: they are the components from which wavefunctions can be built. Structures may be covalent (or non-polar) or ionic (or polar). The representation is frequently kept qualitative so that we speak of important or major contributing structures and minor contributing structures. For example, two major non-equivalent contributing structures for the conjugate base of acetone are
![[resonance arrow]](../greek/RESARR.GIF) H2C--C(CH3)=O
H2C--C(CH3)=OSee also delocalization, Kekulé structure, resonance.
coordinate covalence (coordinate link)
See coordination.
The formation of a covalent bond, the two shared electrons of which have come from only one of the two parts of the molecular entity linked by it, as in the reaction of a Lewis acid and a Lewis base to form a Lewis adduct; alternatively, the bonding formed in this way. In the former sense, it is the reverse of unimolecular heterolysis. "Coordinate covalence" and "coordinate link" are synonymous (obsolescent) terms. The synonym "dative bond" is obsolete.
(The origin of the bonding electrons has by itself no bearing on the character of the bond formed. Thus, the formation of methyl chloride from a methyl cation and a chloride ion involves coordination; the resultant bond obviously differs in no way from the C-Cl bond in methyl chloride formed by any other path, e.g. by colligation of a methyl radical and a chlorine atom.)
The term is also used to describe the number of ligands around a central atom without necessarily implying two-electron bonds.
See also dipolar bond, -adduct.
The coordination number of a specified atom in a chemical species is the number of other atoms directly linked to that specified atom [cf. IUPAC INORGANIC NOMENCLATURE (1990); Rule I-10.2.5]. For example, the coordination number of carbon in methane is four, and it is five in protonated methane, CH5+. (The term is used in a different sense in the crystallographic description of ionic crystals.)
A transition metal complex that has formally 18 outer shell electrons at the central metal atom.
A transition metal complex that possesses fewer ligands than exist in the coordinatively saturated complex. These complexes usually have fewer than 18 outer shell electrons at the central metal atom.
See crown.
The use of empirical correlations relating one body of experimental data to another, with the objective of finding quantitative estimates of the factors underlying the phenomena involved. Correlation analysis in organic chemistry often uses linear free-energy relations for rates or equilibria of reactions, but the term also embraces similar analysis of physical (most commonly spectroscopic) properties and of biological activity. CHAPMAN and SHORTER (1972, 1978). See also Quantitative Structure-Activity Relationships (QSAR).
See cybotactic region.
coupling constant (spin-spin coupling constant), J (SI unit: Hz (NMR))
A quantitative measure for nuclear spin-spin, nuclear-electron (hyperfine coupling) and electron-electron (fine coupling in EPR) coupling in magnetic resonance spectroscopy. The "indirect" or scalar NMR coupling constants are in a first approximation independent of the external magnetic field and are expressed in Hz.
A region of relatively high electron density between nuclei which arises at least partly from sharing of electrons and gives rise to an attractive force and characteristic internuclear distance. See also agostic, coordination, hydrogen bond, multi-centre bond.
A modification of the Bunnett-Olsen equation of the form
where X is the activity function lg(![[gamma]](../greek/GAMMA.GIF) SH+)/SH+) for an arbitrary reference base. The function X is called the excess acidity because it gives a measure of the difference between the acidity of a solution and that of an ideal solution of the same concentration. In practice X = - (Ho + lg[H+]) and m* = 1 -
SH+)/SH+) for an arbitrary reference base. The function X is called the excess acidity because it gives a measure of the difference between the acidity of a solution and that of an ideal solution of the same concentration. In practice X = - (Ho + lg[H+]) and m* = 1 - ![[Phi]](../greek/PPHI.GIF) . COX and YATES (1978, 1981); LUCCHINI, MODENA, SCORRANO, COX and YATES (1982). See also Bunnett-Olsen equations.
. COX and YATES (1978, 1981); LUCCHINI, MODENA, SCORRANO, COX and YATES (1982). See also Bunnett-Olsen equations.
critical micelle concentration (cmc)
There is a relatively small range of concentrations separating the limit below which virtually no micelles are detected and the limit above which virtually all additional surfactant molecules form micelles. Many properties of surfactant solutions, if plotted against the concentration, appear to change at a different rate above and below this range. By extrapolating the loci of such a property above and below this range until they intersect, a value may be obtained known as the critical micellization concentration (critical micelle concentration), symbol cM, abbreviation cmc (or c.m.c.). As values obtained using different properties are not quite identical, the method by which the cmc is determined should be clearly stated. See IUPAC MANUAL APPENDIX II (1972). See also inverted micelle.
In a system XC6H4GY this is conjugation involving the substituent X, the benzene ring, and the side-chain connective-plus-reaction site GY, i.e. either X is a +R group and GY is a -R group, or X is a -R group and GY is a +R group. In Hammett correlations this situation can lead to the need to apply exalted substituent constants + or -, respectively, as in electrophilic or nucleophilic aromatic substitution, respectively. The term "through resonance" is synonymous. Cross conjugation has also been used to describe the interactions occurring in 2-phenylallyl and and similar systems (DEWAR (1969)).
A molecular entity comprising a monocyclic ligand assembly that contains three or more binding sites held together by covalent bonds and capable of binding a guest in a central (or nearly central) position. The adducts formed are sometimes known as "coronates". The best known members of this group are macrocyclic polyethers, such as "18-crown-6", containing several repeating units -CR2-CR2O- (where R is most commonly H), and known as crown ethers. PEDERSEN (1967); CRAM et al. (1986).
See also host.
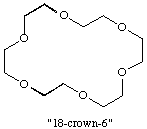
A molecular entity comprising a cyclic or polycyclic assembly of binding sites that contains three or more binding sites held together by covalent bonds, and which defines a molecular cavity in such a way as to bind (and thus "hide" in the cavity) another molecular entity, the guest (a cation, an anion or a neutral species), more strongly than do the separate parts of the assembly (at the same total concentration of binding sites). The adduct thus formed is called a "cryptate". The term is usually restricted to bicyclic or oligocyclic molecular entities.
Example
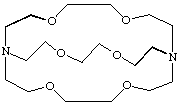
Corresponding monocyclic ligand assemblies (crowns) are sometimes included in this group, if they can be considered to define a cavity in which a guest can hide. The terms "podand" and "spherand" are used for certain specific ligand assemblies. Coplanar cyclic polydentate ligands, such as porphyrins, are not normally regarded as cryptands. DIETRICH, LEHN and SAUVAGE (1969). See also host. For a contribution to the evolving terminology, see also WEBER and VÖGTLE (1980).
In a chemical reaction that yields one product (X) from one conformational isomer (A') and a different product (Y) from another conformational isomer (A") (and provided these two isomers are rapidly interconvertible relative to the rate of product formation, whereas the products do not undergo interconversion) the product composition is not in direct proportion to the relative concentrations of the conformational isomers in the substrate; it is controlled only by the difference in standard free energies (dG) of the respective transition states.
It is also true that the product composition is formally related to the relative concentrations of the conformational isomers A' and A" (i.e. the conformational equilibrium constant) and the respective rate constants of their reactions; these parameters are generally - though not invariably - unknown.
The diagram below represents the energetic situation for transformation of interconverting isomers A and A' into products X and Y.
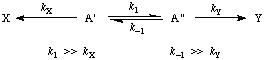
ELIEL (1962); see also SEEMAN et al. (1980), SEEMAN (1983).
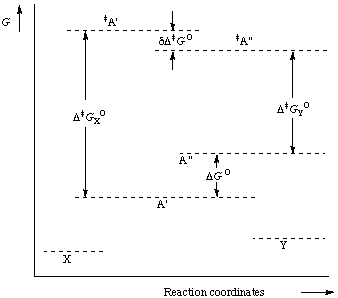
Figure. Curtin-Hammett principle. Transformation of interconverting isomers A' and A" into products X and Y.
That part of a solution in the vicinity of a solute molecule in which the ordering of the solvent molecules is modified by the presence of the solute molecule. The term solvent "cosphere" of the solute has also been used. KOSOWER (1968); STEWART and MORROW (1927). See also solvation.
Formation of a ring compound from a chain by formation of a new bond. See also annulation.
A reaction in which two or more unsaturated molecules (or parts of the same molecule) combine with the formation of a cyclic adduct in which there is a net reduction of the bond multiplicity.
The following two systems of notations have been used for the more detailed specification of cycloadditions, of which the second, more recent system (described under (2)) is preferred:
(1) An (i+j+...) cycloaddition is a reaction in which two or more molecules (or parts of the same molecule), respectively, provide units of i, j,... linearly connected atoms: these units become joined at their respective termini by new sigma bonds so as to form a cycle containing (i+j+...) atoms. In this notation, (a) a Diels-Alder reaction is a (4+2) cycloaddition, (b) the initial reaction of ozone with an alkene is a (3+2) cycloaddition, and (c) the reaction shown below is a (2+2+2) cycloaddition. (N.B.: parentheses (...) are used in the description based on numbers of atoms.)
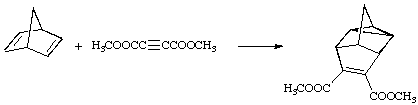
(2) The symbolism [i+j+...] for a cycloaddition identifies the numbers i, j,... of electrons in the interacting units that participate in the transformation of reactants to products. In this notation the reaction (a) and (b) of the preceding paragraph would both be described as [2+4] cycloadditions, and (c) as a [2+2+2] cycloaddition. The symbol a or s (a = antarafacial, s = suprafacial) is often added (usually as a subscript after the number to designate the stereochemistry of addition to each fragment. A subscript specifying the orbitals, viz., , (sigma, pi) with their usual significance) or n (for an orbital associated with a single atom only), may be added as a subscript before the number. Thus the normal Diels-Alder reaction is a [4s+2s] or [4s + 2s] cycloaddition, whilst the reaction
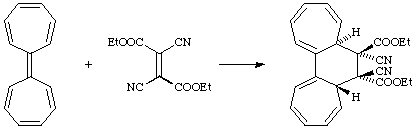
would be a [14a+2s] or [14a + 2s] cycloaddition. (N.B. Square brackets [...] are used in the descriptions based on numbers of electrons.)
Cycloadditions may be pericyclic reactions or (non-concerted) stepwise reactions. The term "dipolar cycloaddition" is used for cycloadditions of 1,3-dipolar compounds. HUISGEN (1968); HUISGEN, GRASHEY and SAUER (1964); WOODWARD and HOFFMANN (1969). See also cheletropic reactions.
The reverse of cycloaddition. The term is preferred to the synonyms "cycloreversion", "retro-addition", and "retrocycloaddition".
See cycloelimination.
CONNOR, K. A. (1991), "Chemical Kinetics", VCH, New York, p. 368.
COX, R. A., and YATES, K. (1978), J. Am. Chem. Soc., 100, 3861-3867.
COX, R. A., and YATES, K. (1981), Can. J. Chem., 59, 2116-2124.
DEWAR, M. J. S. (1969), "The Molecular Orbital Theory for Organic Chemistry", McGraw-Hill, New York.
DIETRICH, B., LEHN, J. M., and SAUVAGE, J. P. (1969), Tetrahedron Lett., 2889-2892.
ELIEL, E. L. (1962), "Stereochemistry of Carbon Compounds", McGraw Hill, New York.
HARTMANNS, J., KLENKE, K., and METZGER, J. O. (1986), Chem. Ber., 119, 488-499.
HUISGEN, R. (1968), Angew. Chem., Int. Ed. Engl., 7, 321-328.
*IUPAC STEREOCHEMICAL TERMINOLOGY (1993). IUPAC: Organic Chemistry Division: Basic Terminology of Stereochemistry. IDCNS and public review. Now published as Basic Terminology of Stereochemistry (IUPAC Recommendations 1996) in Pure Appl. Chem., 68, 2193-2222 (1996).
KOSOWER, E. M. (1968), "An Introduction to Physical Organic Chemistry". Wiley, New York.
PEDERSEN, C. J. (1967), J. Am. Chem. Soc., 89, 7017-7036.
SEEMAN, J. I. (1983), Chem. Rev., 83, 83-134.
STEWART, G. W., and MORROW, R. M. (1927), Proc. Natl. Acad. Sci. USA, 13, 222-223.
WEBER, A., and VÖGTLE, F. (1980), Inorg. Chim. Acta, 45, L65-L67.
WOODWARD, R. B., and HOFFMANN, R. (1969), Angew. Chem., Int. Ed. Engl., 8, 781-853.
Return to home page for Glossary of terms used in Physical Organic Chemistry.